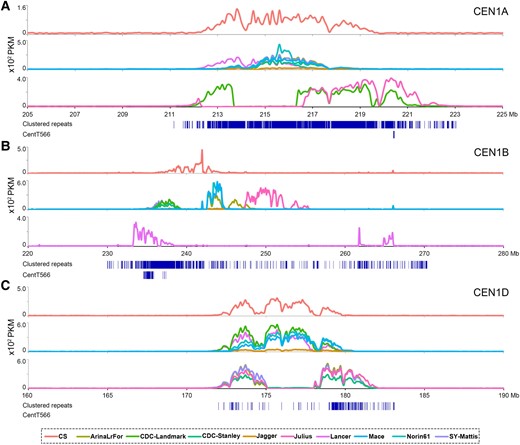
Telomere-to-telomere genome assembly reveals the genomic architecture of disease resistance and yield coordination in elite wheat YM33
Authors: Guofeng Lv,Yating Wang,Heping Zhang,Yuning Shen,Wenjing Hu,Datong Liu,Mengmeng Liu,Wenna Wang,Yuwen Gao,Caixia Lan,Tongde Bie,Hongya Wu,Wei Chen,Yong Zhang,Jianwei Zhang,Chao He,Wenhao Yan,Derong Gao
Journal: Molecular Plant
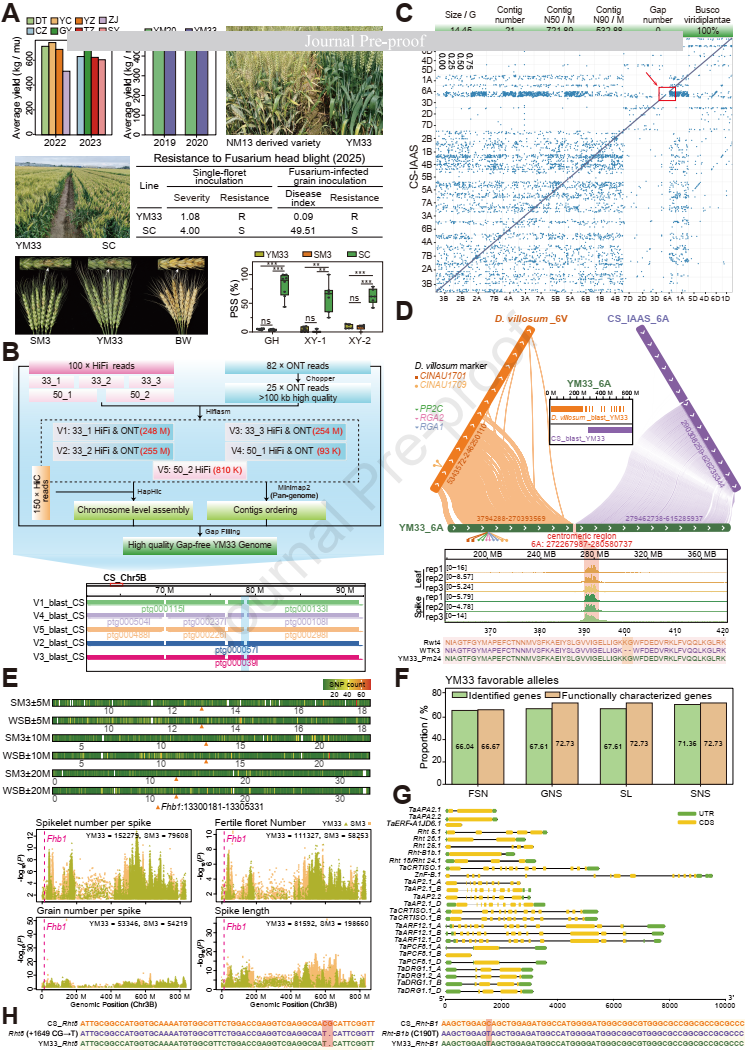
Wheat (Triticum aestivum) faces significant threats from diseases such as powdery mildew (Blumeria graminis) and Fusarium head blight (FHB; caused by Fusarium graminearum), which cause severe yield losses. Moreover, the antagonism between yield-related traits and disease resistance makes yield resistance coordination a major challenge in wheat breeding. The lack of genetic resources combining both disease resistance and high yield constrains the elucidation of underlying resistance-yield trade-off mechanisms, thereby hindering the development of high-yield and disease-resistant wheat cultivars. Remarkably, Yangmai 33 (YM33), a notable wheat cultivar with resistance to both powdery mildew and FHB as well as high-yield performance, was recently developed. It offers a unique opportunity to dissect the genomic architecture underlying the coordination between disease resistance and yield.
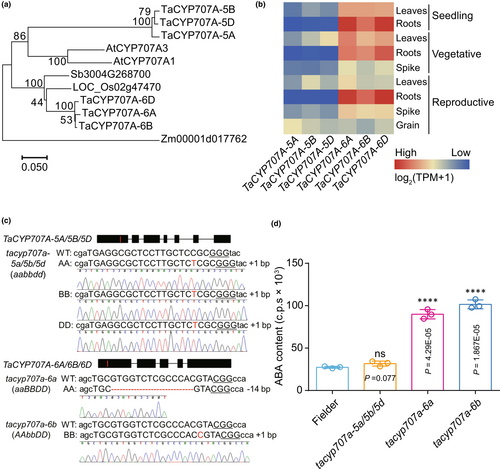
Modulation of an evolutionarily conserved epigenetic regulon controlling abscisic acid catabolism enhances drought tolerance in wheat
Authors: Yunzhen Li, Liujie Jin, Wanying Li, Ke Wang, Handong Su, Hailiang Mao, Wei Chen, Caixia Lan, Qiang Li, Kerstin Kaufmann, Wenhao Yan
Journal: New Phytologist
Drought stress significantly reduces crop yield by triggering abscisic acid (ABA) accumulation in plants. It involves the suppression of CYP707A genes, which encode enzymes that catalyze ABA. However, little is known about epigenetic control in the CYP707A gene-mediated drought stress response in wheat. In this study, we reported that TaCYP707A-6A/6B/6D but not TaCYP707A-5A/5B/5D participates in drought response in common wheat. Disruption of TaCYP707A-6B showed enhanced drought tolerance but also decreased fertility. Expression of TaCYP707A-6B is negatively associated with H3K27me3 level. An evolutionarily conserved CTCTGYTY motif cluster (binding site for a Jumonji H3K27me3 demethylase) was found in the intron of TaCYP707A-6B as well as the intron of CYP707A homologs in other plant species. Blocking the CTCTGYTY motif by dead Cas9 (dCas9) maintained a high level of H3K27me3 on the CYP707A gene, while decreasing its expression level leading to enhanced drought tolerance in both wheat and Arabidopsis. In particular, the mutant in which the intron bound by H3K27me3 demethylase was cut out without change of splicing pattern showed enhanced drought tolerance. Therefore, our study provides a novel approach to improve plant drought tolerance by manipulating an evolutionarily conserved cis-element bound by histone demethylases in the intron of CYP707A genes.
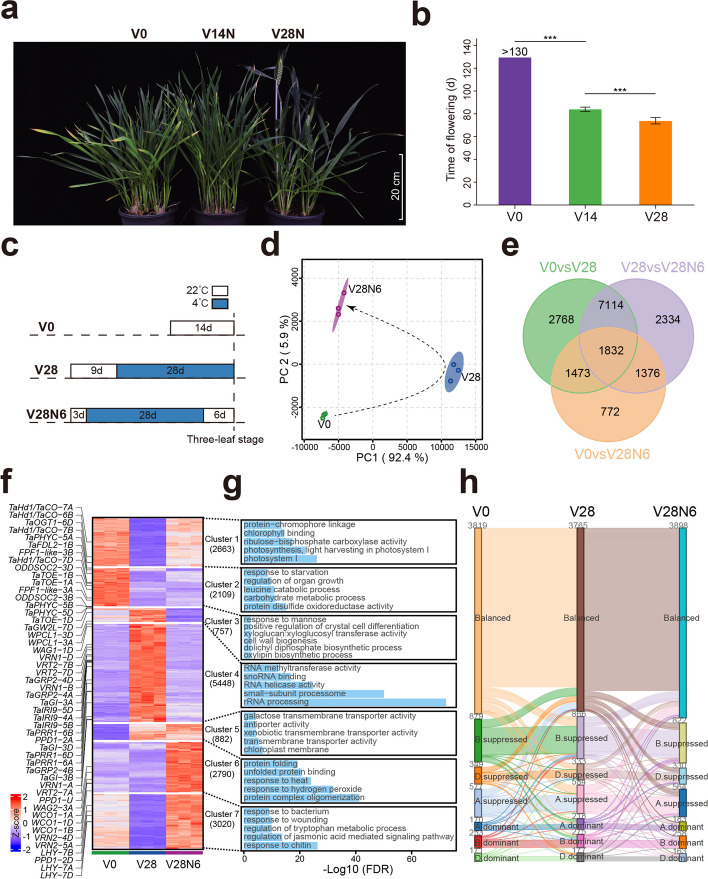
Chromatin loops gather targets of upstream regulators together for efficient gene transcription regulation during vernalization in wheat
Authors: Yanyan Liu, Xintong Xu, Chao He, Liujie Jin, Ziru Zhou, Jie Gao, Minrong Guo, Xin Wang, Chuanye Chen, Mohammed H. Ayaad, Xingwang Li & Wenhao Yan
Journal: Genome Biology
Plants respond to environmental stimuli by altering gene transcription that is highly related with chromatin status, including histone modification, chromatin accessibility, and three-dimensional chromatin interaction. Vernalization is essential for the transition to reproductive growth for winter wheat. How wheat reshapes its chromatin features, especially chromatin interaction during vernalization, remains unknown.
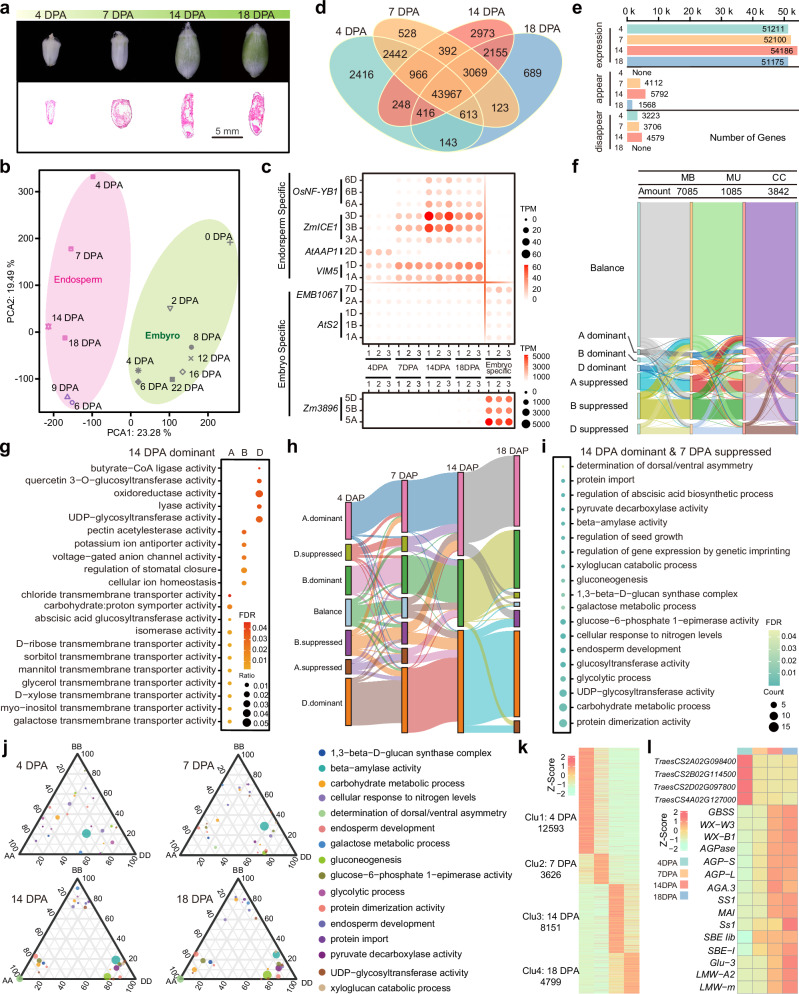
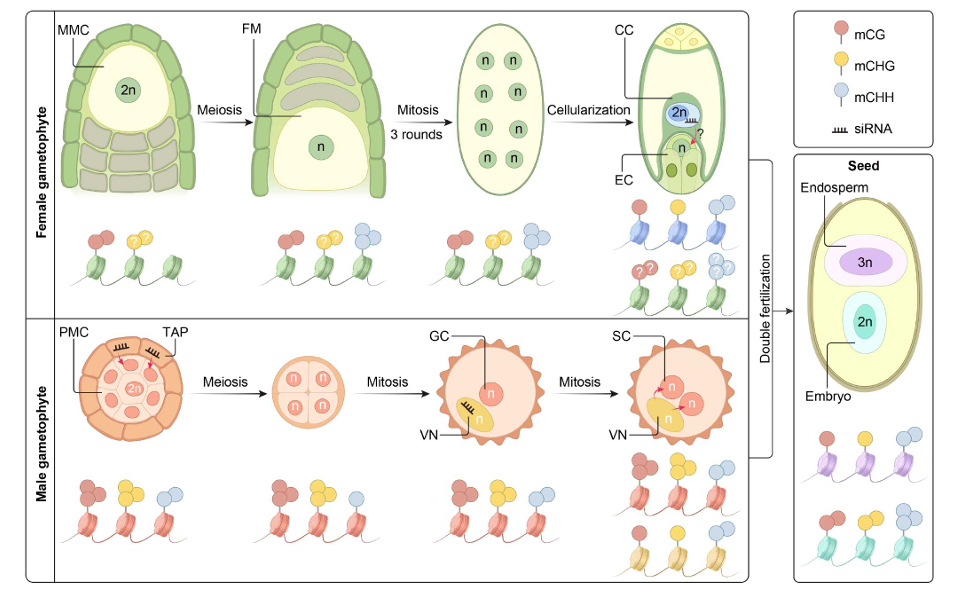
Dynamic atlas of histone modifications and gene regulatory networks in endosperm of bread wheat
Authors: Chao He, Siteng Bi, Yuqi Li, Chengxiang Song, Heping Zhang, Xintong Xu, Qiang Li, Sulaiman Saeed, Wei Chen, Chunjie Zhao, Caixia Lan, Handong Su, Hailiang Mao & Wenhao Yan
Journal: Nature Communications
Dissecting the genetic basis of seed traits in wheat is impeded by limited genetic polymorphisms and significant variations caused by environmental conditions and seed position in a spikelet. Seed performance is largely determined by endosperm development controlled by spatiotemporal variation in gene activities, which is greatly affected by chromatin status. Here, we map genome-wide dynamic distributions of H3K27me3, H3K4me3 and H3K9ac modifications and profile gene transcription across wheat endosperm development. The combinatorial effects of active and repressive marks ensure spatiotemporal dynamic gene expression, especially for starch biosynthesis. By scanning the transcription factor binding motifs in the ATAC-seq peaks, hub regulators are identified from the regulatory network. In addition, we observe significant correlations between sequence polymorphisms of hub regulators and variations in seed traits in a germplasm population. Thus, the analysis of genomic regulatory activities together with genetic variation provides a robust approach to dissect seed traits in bread wheat.
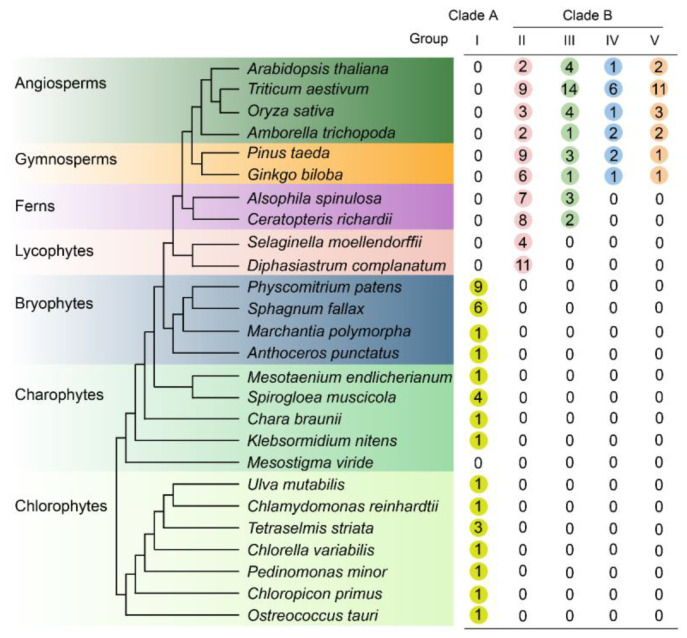
Evolutionary Analysis and Catalytic Function of LOG Proteins in Plants
Authors:Chunjie Zhao, Huanran Yin, Yuqi Li, Jiacheng Zhou, Siteng Bi, Wenhao Yan, Yunzhen Li
Journal: Genes
Our phylogeny showed that LOG proteins could be divided into five groups and revealed three major duplication events giving rise to four distinct groups of vascular LOG proteins. LOG proteins share a conserved structure characterized by a canonical motif arrangement comprising motifs 1, 2, 3, 4, 5, 6, and 7. Two significant changes in LOG motif composition occurred during the transition to land plants: the emergence of motif 3 in charophyte LOG sequences and the subsequent acquisition of motif 8 at the C-terminus of LOG proteins. Enzymatic assays demonstrated that LOG proteins can be classified into two groups based on their enzyme activity. One group act as cytokinin riboside 5'-monophosphate phosphoribohydrolase and primarily convert iPRMP to iP, while the other group act as 5'-ribonucleotide phosphohydrolase, and preferentially produce iPR from the same substrates. TaLOG5-4A1, TaLOG5-4A2, TaLOG5-5B2, and TaLOG5-D1 shared conserved residues in the critical motif and were predicted to have similar protein structures, but displayed distinct enzymatic activities.
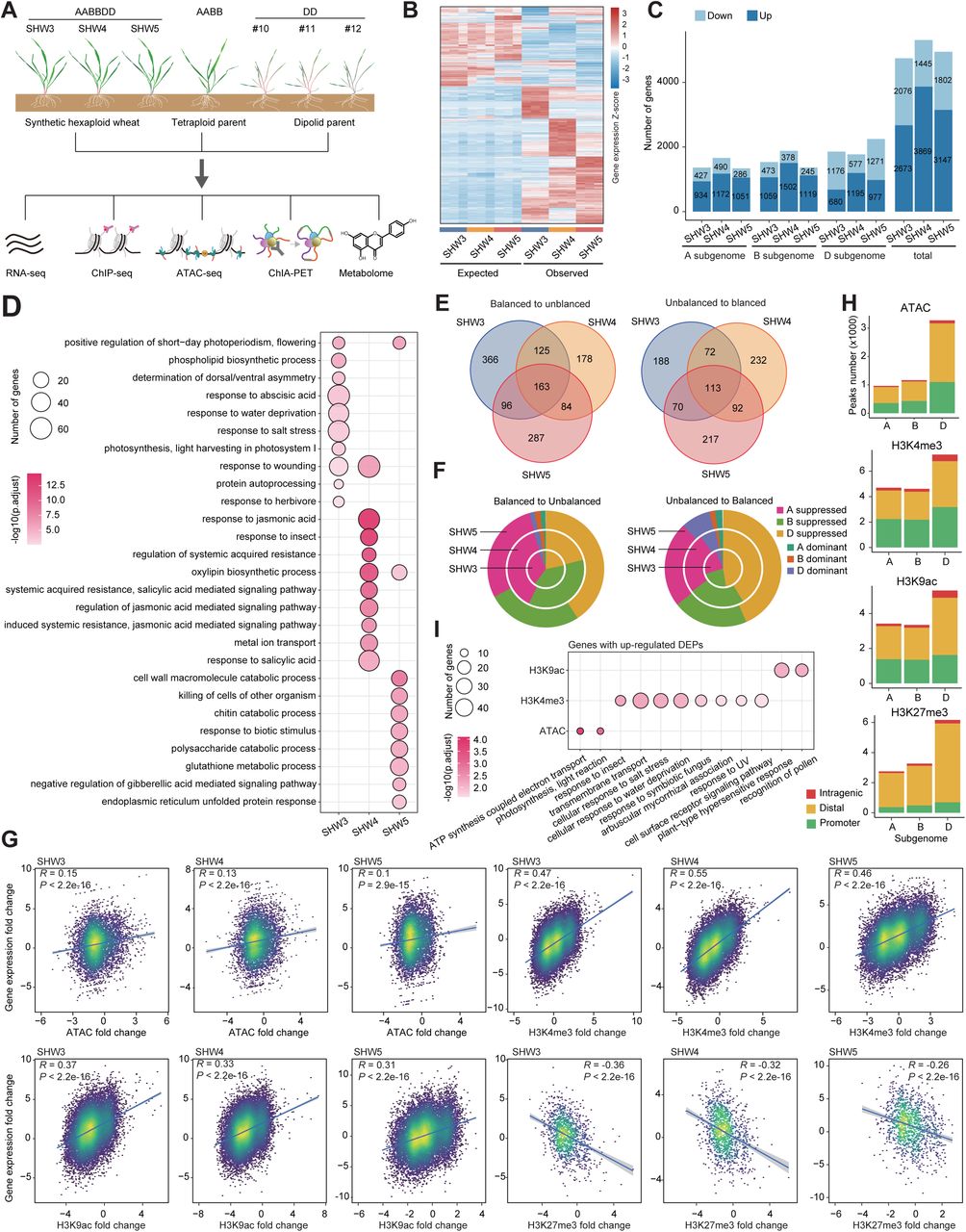
Integration of the DD-genome reshapes gene transcription, chromatin architecture and metabolome of allohexaploid wheat leading to enhanced adaptability
Authors: Yanyan Liu, Tao Zhu, Xinkai Zhou, Wei Chen, Chao He, Xin Wang, Chuanye Chen, Jiaqi Wei, Caixia Lan, Mengmeng Liu, Handong Su, Qiang Li, Xin Hu, Siteng Bi, Weizhi Ouyang, Xingwang Li, Hailiang Mao, Masahiro Kishi, Kerstin Kaufmann, Alisdair R. Fernie, Dijun Chen, Wenhao Yan
The integration, through hybridization, of the DD genome into domesticated tetraploid wheat gave rise to allohexaploid wheat, the most cultivated wheat globally growing across diverse environmental conditions. However, the regulatory basis of this integration on increased environmental adaptability in allohexaploid remains largely unexplored. Here, we investigated the change of transcriptome, epigenome as well as the chromatin interactome, and metabolome in three independent polyploidization events representing DD genome integration. Our findings reveal that polyploidization events induce the activation of defense-related genes through comprehensive reorganization of epigenome and chromatin architecture. DD integration not only brings an additional gene copy but also activates the homoeologs existing in the A and B subgenomes through chromatin interactions. Furthermore, secondary metabolites represented by alkaloids and flavonoids that are crucial for environmental adaptation, are significantly enriched following polyploidization. Thus, hexaploid wheat exhibits enhanced tolerance to alkalinity, UV-B light stress and high salt conditions was observed. These results highlight the indispensable role of DD genome integration in the adaptability of allohexaploid wheat during its evolution.
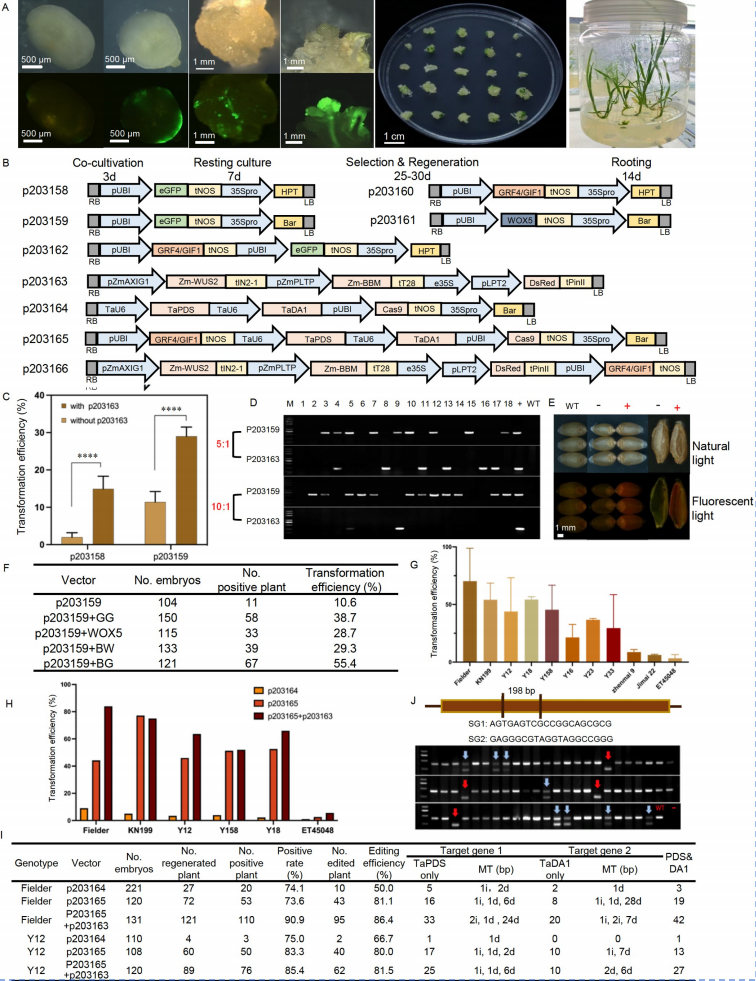
Overcoming genotypic dependency and bypassing immature embryos in wheat transformation by using morphogenic regulators
Authors: Ziru Zhou, Yawen Yang, Guo Ai, Miaomiao Zhao, Baozhu Han, Chunjie Zhao, Yiqian Chen, Yuwei Zhang, Hong Pan, Caixia Lan, Chao He, Qiang Li, Jieting Xu, Wenhao Yan
Journal: Science China Life Sciences
All the results strongly support that the combination of BBM-WUS and GRF/GIF not only promotes transformation and gene editing efficiency in wheat across different genotypes, but also allows to skip the need of immature embryos for wheat transformation.
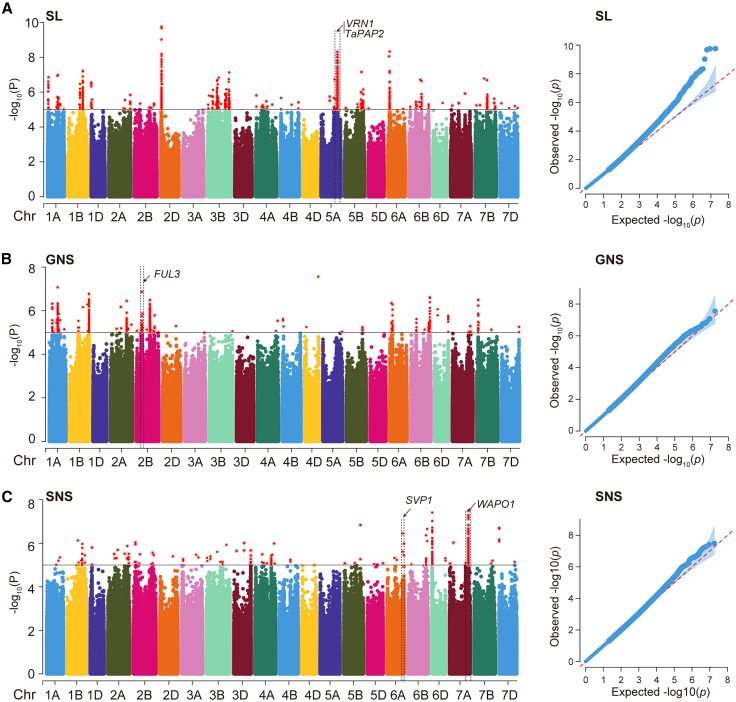
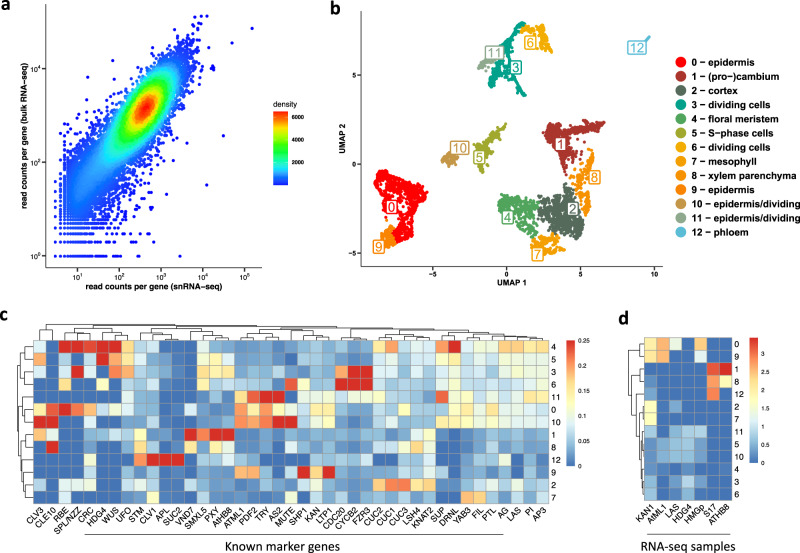
Dissecting the molecular basis of spike traits by integrating gene regulatory networks and genetic variation in wheat
Authors: Guo Ai, Chao He, Siteng Bi, Ziru Zhou, Ankui Liu, Xin Hu, Yanyan Liu, Liujie Jin, JiaCheng Zhou, Heping Zhang, Dengxiang Du, Hao Chen, Xin Gong, Sulaiman Saeed, Handong Su, Caixia Lan, Wei Chen, Qiang Li, Hailiang Mao, Lin Li, Hao Liu, Dijun Chen, Kerstin Kaufmann, Khaled F Alazab, Wenhao Yan
Journal: Plant Communications
Spike architecture influences both grain weight and grain number per spike, which are the two major components of grain yield in bread wheat (Triticum aestivum L.). However, the complex wheat genome and the influence of various environmental factors pose challenges in mapping the causal genes that affect spike traits. Here, we systematically identified genes involved in spike trait formation by integrating information on genomic variation and gene regulatory networks controlling young spike development in wheat. We identified 170 loci that are responsible for variations in spike length, spikelet number per spike, and grain number per spike through genome-wide association study and meta-QTL analyses. We constructed gene regulatory networks for young inflorescences at the double ridge stage and the floret primordium stage, in which the spikelet meristem and the floret meristem are predominant, respectively, by integrating transcriptome, histone modification, chromatin accessibility, eQTL, and protein-protein interactome data. From these networks, we identified 169 hub genes located in 76 of the 170 QTL regions whose polymorphisms are significantly associated with variation in spike traits. The functions of TaZF-B1, VRT-B2, and TaSPL15-A/D in establishment of wheat spike architecture were verified. This study provides valuable molecular resources for understanding spike traits and demonstrates that combining genetic analysis and developmental regulatory networks is a robust approach for dissection of complex traits.
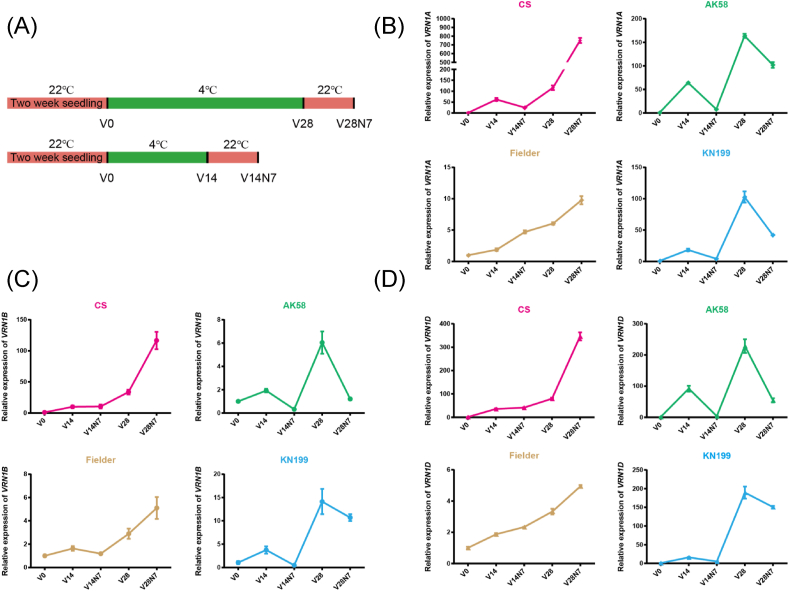
Epigenetic control on transcription of vernalization genes and whole-genome gene expression profile induced by vernalization in common wheat
Authors:Yunzhen Li, Liujie Jin, Xinyu Liu, Chao He, Siteng Bi, Sulaiman Saeed, Wenhao Yan
Journal: Plant Diversity
Vernalization is necessary for winter wheat to flower. However, it is unclear whether vernalization is also required for spring wheat, which is frequently sown in fall, and what molecular mechanisms underlie the vernalization response in wheat varieties. In this study, we examined the molecular mechanisms that regulate vernalization response in winter and spring wheat varieties. For this purpose, we determined how major vernalization genes (VRN1, VRN2, and VRN3) respond to vernalization in these varieties and whether modifications to histones play a role in changes in gene expression. We also identified genes that are differentially regulated in response to vernalization in winter and spring wheat varieties. We found that in winter wheat, but not in spring wheat, VRN1 expression decreases when returned to warm temperature following vernalization. This finding may be associated with differences between spring and winter wheat in the levels of tri-methylation of lysine 27 on histone H3 (H3K27me3) and tri-methylation of lysine 4 on histone H3 (H3K4me3) at the VRN1 gene. Analysis of winter wheat transcriptomes before and after vernalization revealed that vernalization influences the expression of several genes, including those involved in leucine catabolism, cysteine biosynthesis, and flavonoid biosynthesis. These findings provide new candidates for further study on the mechanism of vernalization regulation in wheat.
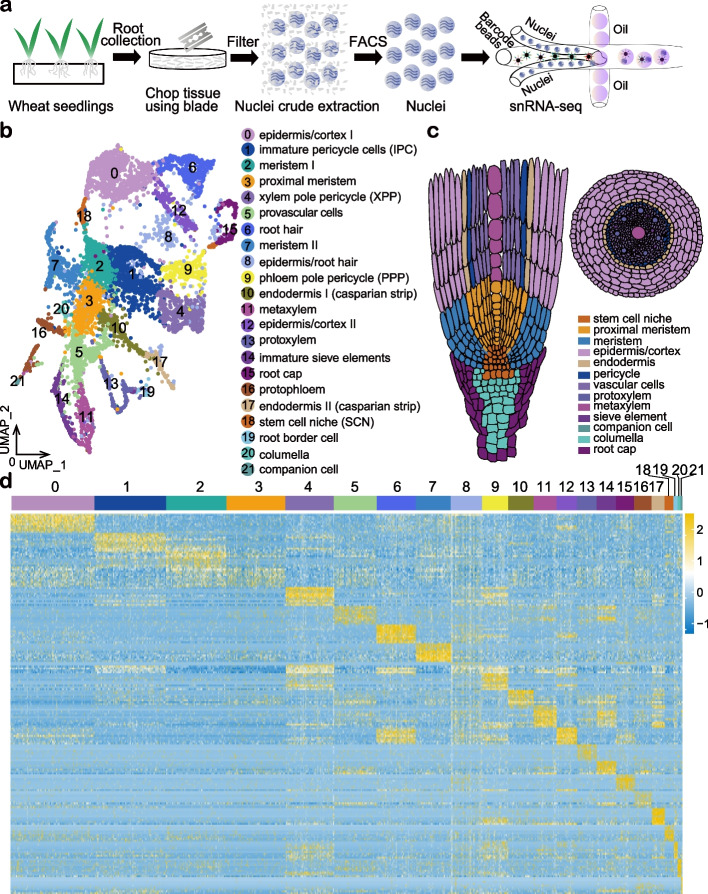
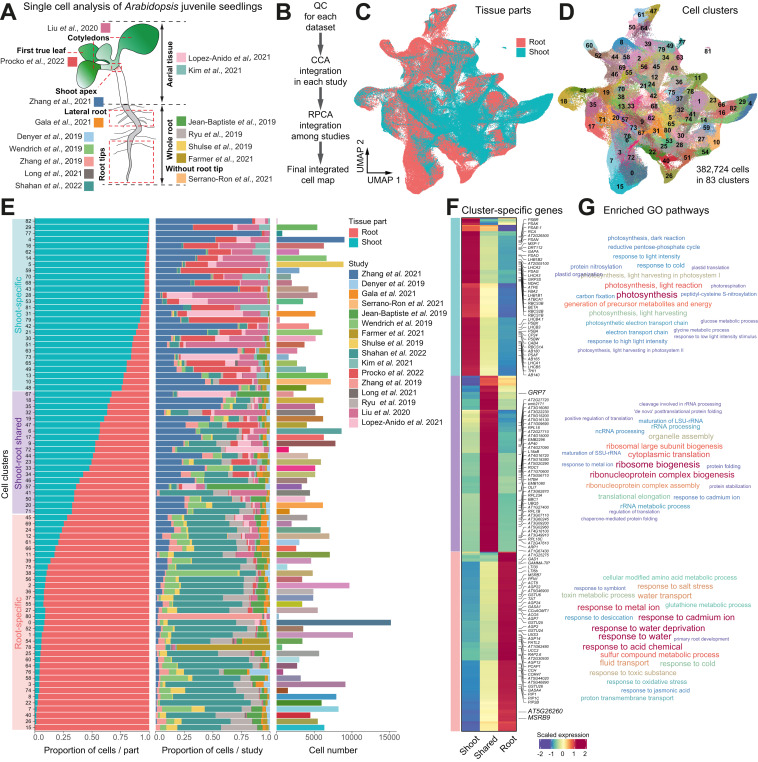
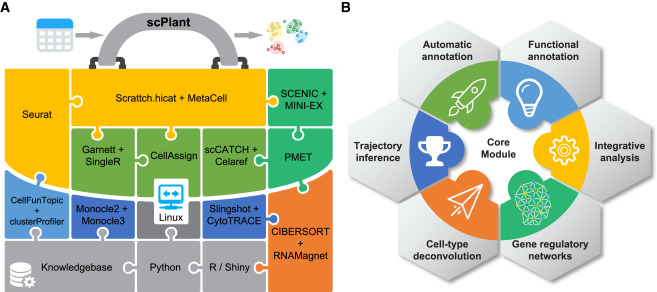
Asymmetric gene expression and cell-type-specific regulatory networks in the root of bread wheat revealed by single-cell multiomics analysis
Authors:Lihua Zhang, Chao He , Yuting Lai, Yating Wang, Lu Kang, Ankui Liu, Caixia Lan, Handong Su, Yuwen Gao, Zeqing Li, Fang Yang, Qiang Li, Hailiang Mao, Dijun Chen, Wei Chen, Kerstin Kaufmann, Wenhao Yan
Journal: Genome Biology
Here, we perform single nuclei RNA sequencing and ATAC sequencing of wheat root to study the asymmetric gene transcription, reconstruct cell differentiation trajectories and cell-type-specific gene regulatory networks. We identify 22 cell types. We then reconstruct cell differentiation trajectories that suggest different origins between epidermis/cortex and endodermis, distinguishing bread wheat from Arabidopsis. We show that the ratio of asymmetrically transcribed triads varies greatly when analyzing at the single-cell level. Hub transcription factors determining cell type identity are also identified. In particular, we demonstrate that TaSPL14 participates in vasculature development by regulating the expression of BAM1. Combining single-cell transcription and chromatin accessibility data, we construct the pseudo-time regulatory network driving root hair differentiation. We find MYB3R4, REF6, HDG1, and GATAs as key regulators in this process.
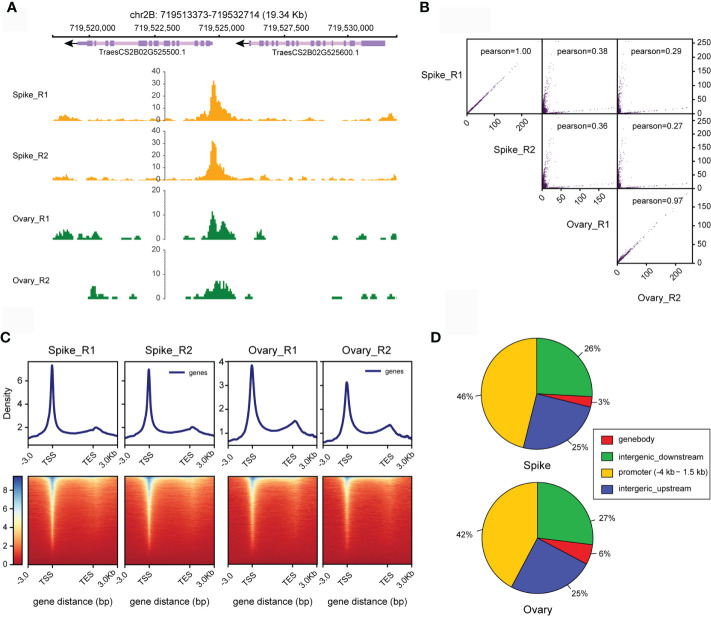
Mapping open chromatin by ATAC-seq in bread wheat
Authors:Xin Wang, Chuanye Chen, Chao He, Dijun Chen, Wenhao Yan
Journal: Frontiers in Plant Science
Gene transcription is largely regulated by cis-regulatory elements. Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) is an emerging technology that can accurately map cis-regulatory elements in animals and plants. However, the presence of cell walls and chloroplasts in plants hinders the extraction of high-quality nuclei, thereby affects the quality of ATAC-seq data. Meanwhile, it is tricky to perform ATAC-seq with different tissue types, especially for those with limited size and amount. Moreover, with rapid growth of ATAC-seq datasets from plants, powerful and easy-to-use data analysis pipelines for ATAC-seq, especially for wheat is lacking. Here, we provided an all-in-one solution for mapping open chromatin in wheat including both experimental and data analysis procedure. We efficiently obtained nuclei with less cell debris from various wheat tissues. High-quality ATAC-seq data from young spike and ovary, which are hard to harvest were generated. We determined that the saturation sequencing depth of wheat ATAC-seq is about 16 Gb. Particularly, we developed a powerful and easy-to-use online pipeline to analyze the wheat ATAC-seq data and this pipeline can be easily extended to other plant species. The method developed here will facilitate plant regulatory genome study not only for wheat but also for other plant species.
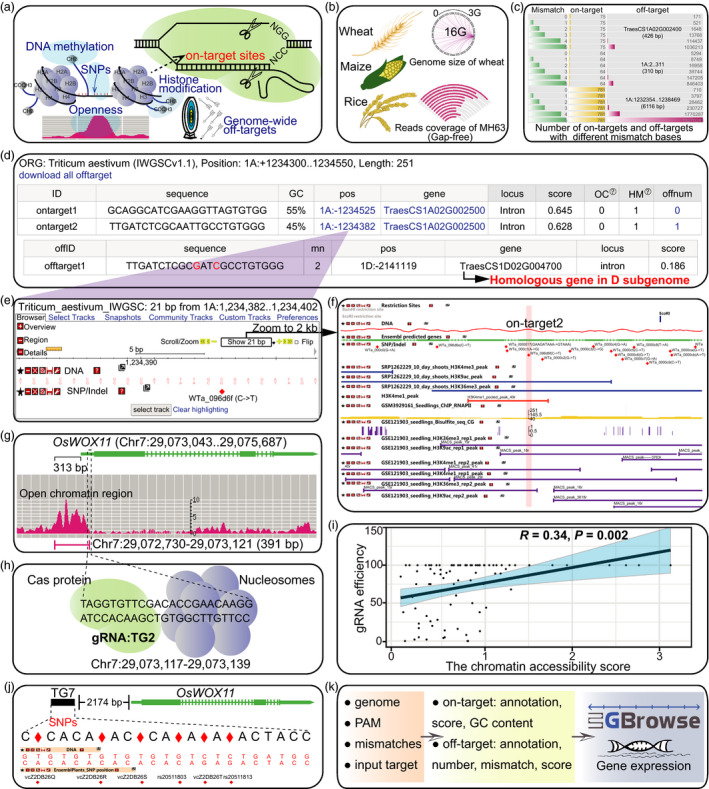
CRISPR-Cereal: a guide RNA design tool integrating regulome and genomic variation for wheat, maize and rice
Authors:Chao He, Hao Liu, Dijun Chen, Wen-Zhao Xie, Mengxin Wang, Yuqi Li, Xin Gong, Wenhao Yan, Ling-Ling Chen
we developed CRISPR-Cereal, a web-based gRNA design tool integrates the information of gene expression profile, chromatin status including chromatin openness and histone modifications, and SNP variations of the putative targets for three leading crops, wheat (Triticum aestivum), maize (Zea mays) and rice (Orazy sativa)
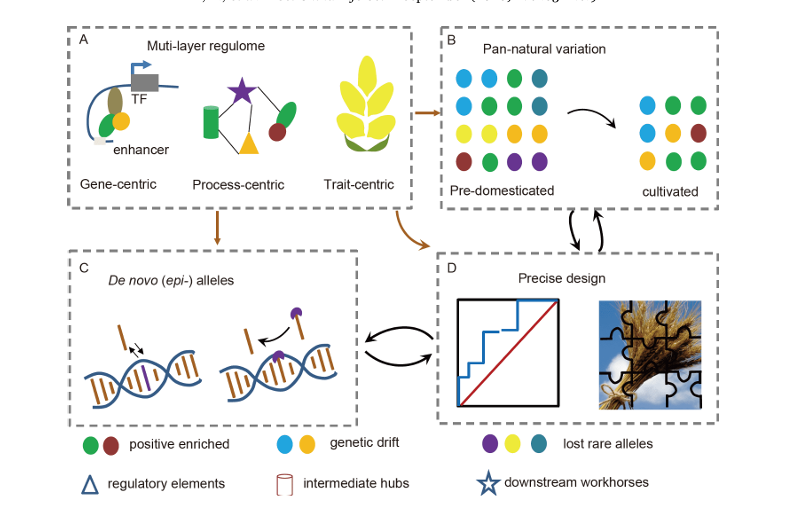
Exploitation of genetic resources based on regulome and gene editing in crops
Authors:Yunzhen Li , Wenhao Yan
Journal: Science China Life Sciences
Crop traits are governed by complex gene networks, and by analyzing regulatory elements and constructing such networks, new functional sites can be identified; with precise editing tools like CRISPR, it is possible not only to restore favorable variations lost during domestication but also to create novel alleles absent in nature, thereby advancing future molecular breeding and crop improvement.
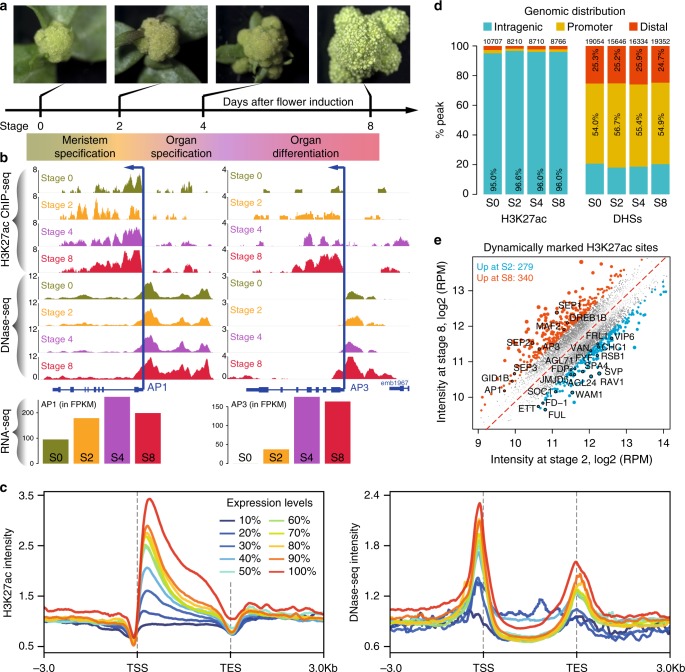
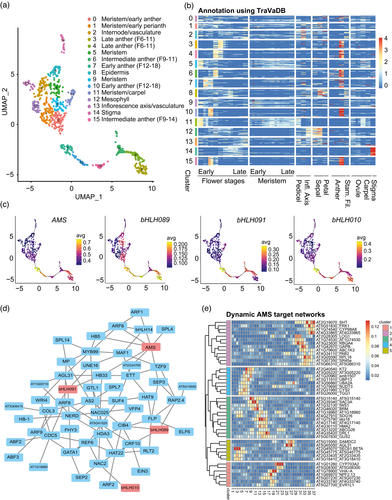
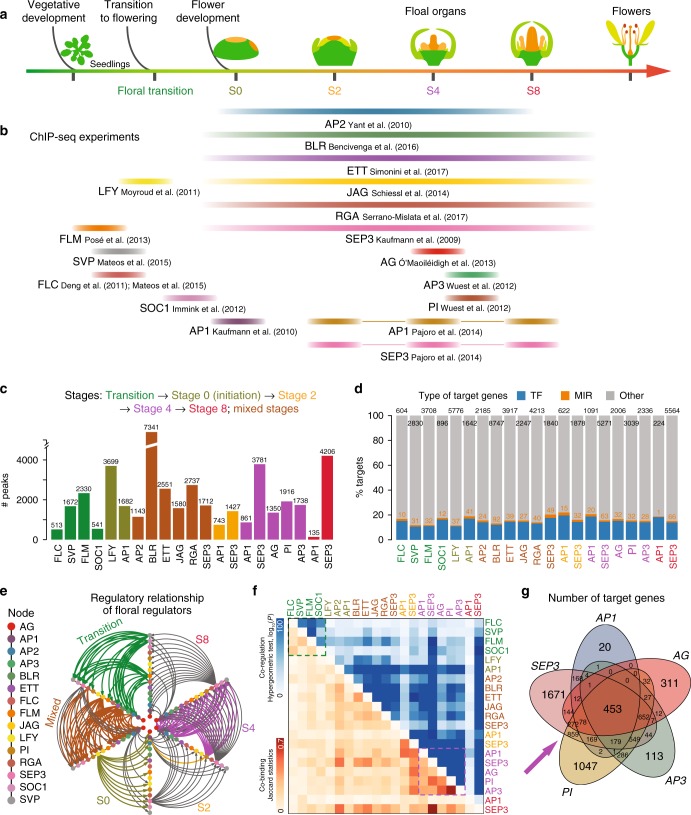
Dynamic control of enhancer activity drives stage-specific gene expression during flower morphogenesis
Authors:Wenhao Yan, Dijun Chen, Julia Schumacher, Diego Durantini, Julia Engelhorn, Ming Chen, Cristel C Carles, Kerstin Kaufmann
Journal: Nature Communications
Enhancers are critical for developmental stage-specific gene expression, but their dynamic regulation in plants remains poorly understood. Here we compare genome-wide localization of H3K27ac, chromatin accessibility and transcriptomic changes during flower development in Arabidopsis. H3K27ac prevalently marks promoter-proximal regions, suggesting that H3K27ac is not a hallmark for enhancers in Arabidopsis. We provide computational and experimental evidence to confirm that distal DNase І hypersensitive sites are predictive of enhancers. The predicted enhancers are highly stage-specific across flower development, significantly associated with SNPs for flowering-related phenotypes, and conserved across crucifer species. Through the integration of genome-wide transcription factor (TF) binding datasets, we find that floral master regulators and stage-specific TFs are largely enriched at developmentally dynamic enhancers. Finally, we show that enhancer clusters and intronic enhancers significantly associate with stage-specific gene regulation by floral master TFs. Our study provides insights into the functional flexibility of enhancers during plant development, as well as hints to annotate plant enhancers.
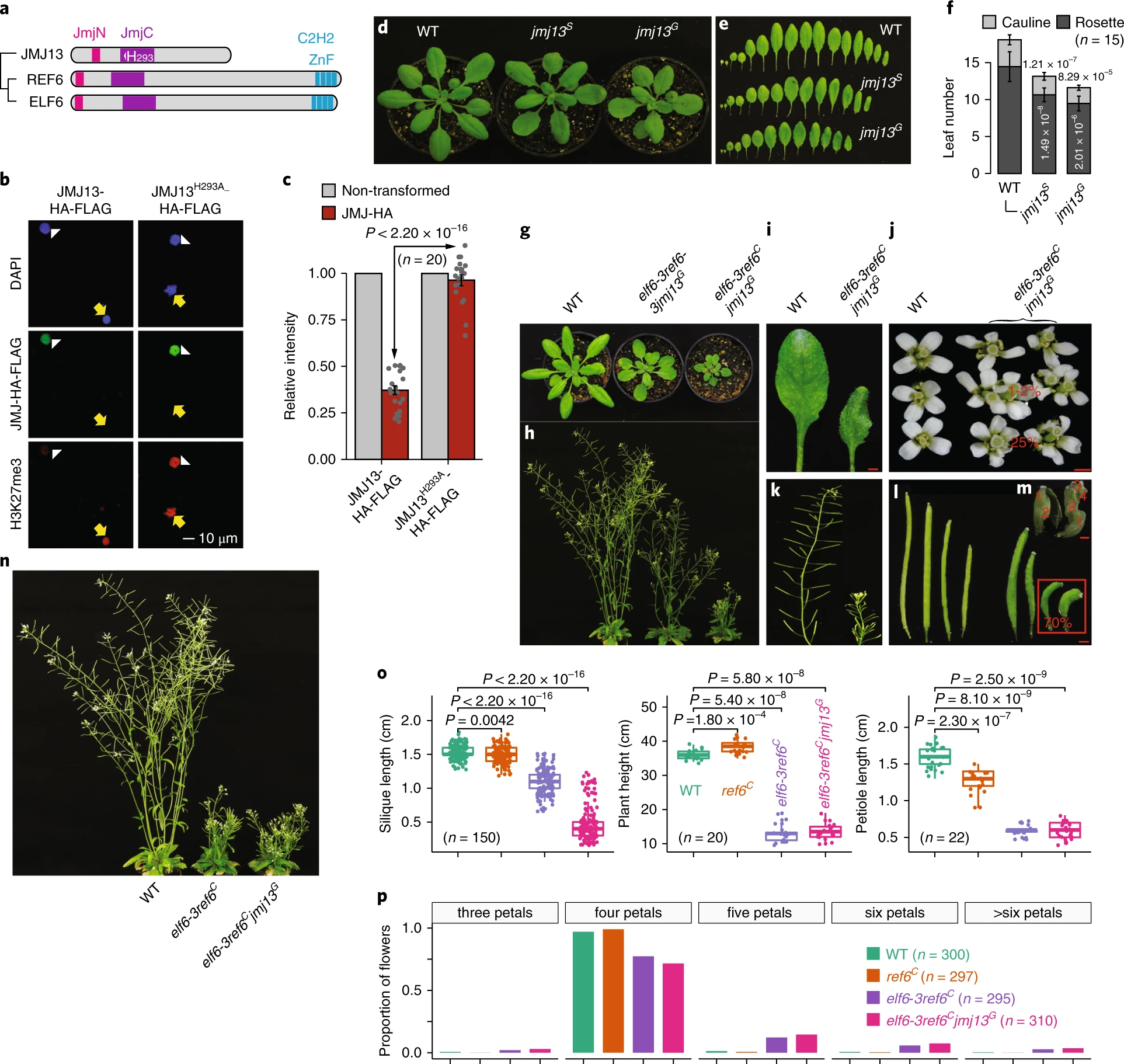
Dynamic and spatial restriction of Polycomb activity by plant histone demethylases
Authors:Wenhao Yan, Dijun Chen, Cezary Smaczniak, Julia Engelhorn, Haiyang Liu, Wenjing Yang, Alexander Graf, Cristel C. Carles, Dao-Xiu Zhou & Kerstin Kaufmann
Journal: Nature Plants
Targeted changes in chromatin state at thousands of genes are central to eukaryotic development. RELATIVE OF EARLY FLOWERING 6 (REF6) is a Jumonji-type histone demethylase that counteracts Polycomb repressive complex 2 (PRC2)-mediated gene silencing in plants and was reported to select its binding sites in a direct, sequence-specific manner1,2,3. Here we show that REF6 and its two close paralogues determine spatial ‘boundaries’ of the repressive histone H3K27me3 mark in the genome and control the tissue-specific release from PRC2-mediated gene repression. Targeted mutagenesis revealed that these histone demethylases display pleiotropic, redundant functions in plant development, several of which depend on trans factor-mediated recruitment. Thus, Jumonji-type histone demethylases restrict repressive chromatin domains and contribute to tissue-specific gene activation via complementary targeting mechanisms.
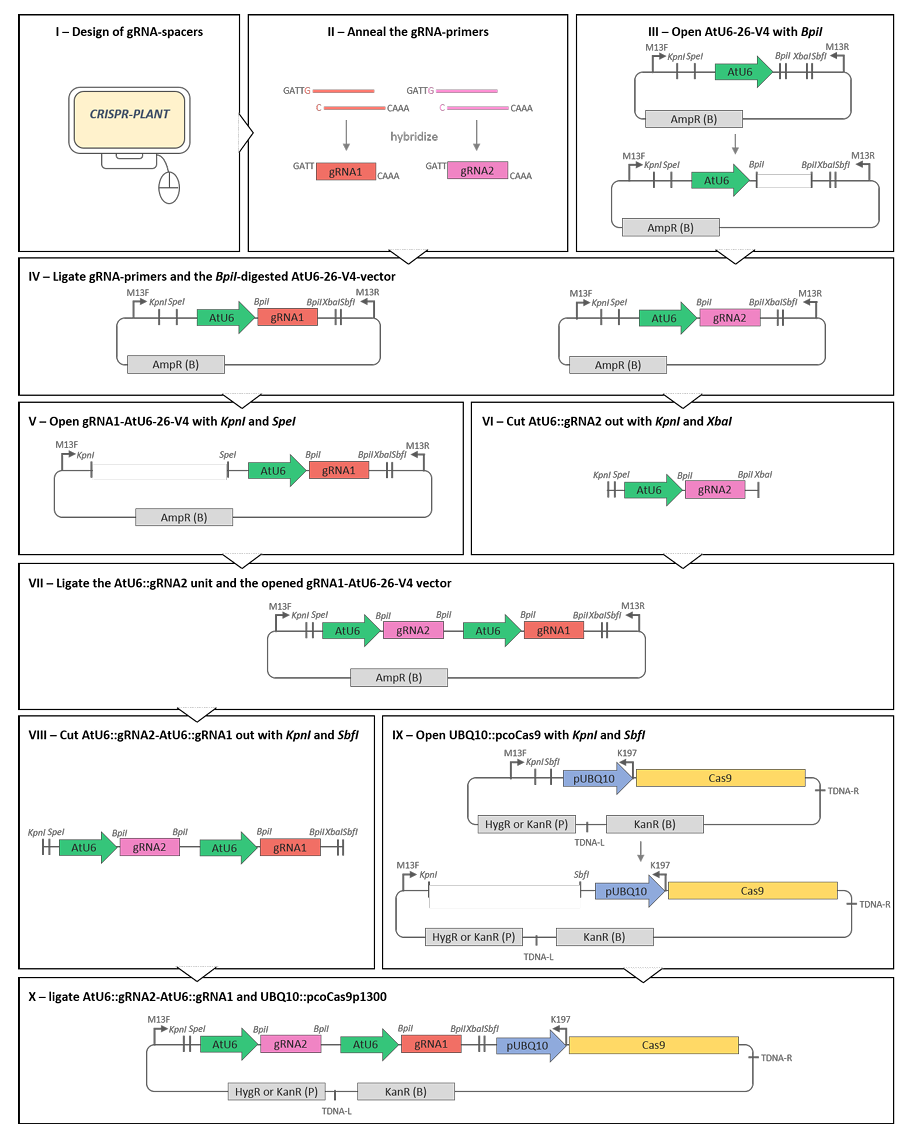
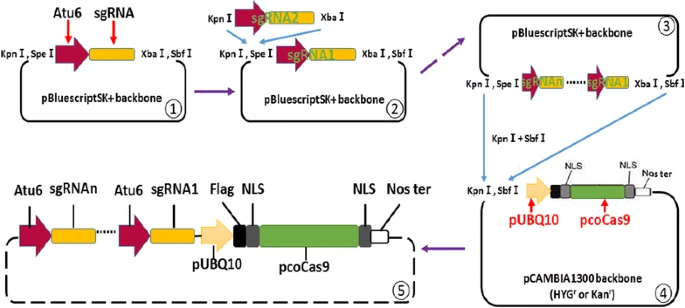
Multiplexed GuideRNA-expression to Efficiently Mutagenize Multiple Loci in Arabidopsis by CRISPR-Cas9
Authors:Julia Schumacher, Kerstin Kaufmann and Wenhao Yan
Journal: Bio-Protocol
Since the discovery of the CRISPR (clustered regularly interspaced short palindromic repeats)-associated protein (Cas) as an efficient tool for genome editing in plants (Li et al., 2013; Shan et al., 2013; Nekrasov et al., 2013), a large variety of applications, such as gene knock-out, knock-in or transcriptional regulation, has been published. So far, the generation of multiple mutants in plants involved tedious crossing or mutagenesis followed by time-consuming screening of huge populations and the use of the Cas9-system appeared a promising method to overcome these issues. We designed a binary vector that combines both the coding sequence of the codon optimized Streptococcus pyogenes Cas9 nuclease under the control of the Arabidopsis thaliana UBIQUITIN10 (UBQ10)-promoter and guideRNA (gRNA) expression cassettes driven by the A. thaliana U6-promoter for efficient multiplex editing in Arabidopsis (Yan et al., 2016). Here, we describe a step-by-step protocol to cost-efficiently generate the binary vector containing multiple gRNAs and the Cas9 nuclease based on classic cloning procedure.
Efficient multiplex mutagenesis by RNA-guided Cas9 and its use in the characterization of regulatory elements in the AGAMOUS gene
Authors:Wenhao Yan, Dijun Chen, Kerstin Kaufmann
Journal: Plant Methods
Here, an RNA-guided Cas9 system was optimized to enable efficient multiplex editing in Arabidopsis thaliana. We demonstrate the flexibility of our system for knockout of multiple genes, and to generate heritable large-fragment deletions in the genome. As a proof of concept, the function of part of the second intron of the flower development gene AGAMOUS in Arabidopsis was studied by generating a Cas9-free mutant plant line in which part of this intron was removed from the genome. Further analysis revealed that deletion of this intron fragment results 40 % decrease of AGAMOUS gene expression without changing the splicing of the gene which indicates that this regulatory region functions as an activator of AGAMOUS gene expression.

Molecular mechanisms of floral organ specification by MADS domain proteins
Authors:Wenhao Yan, Dijun Chen, Kerstin Kaufmann
Journal: FEBS Letters
Flower development is a model system to understand organ specification in plants. The identities of different types of floral organs are specified by homeotic MADS transcription factors that interact in a combinatorial fashion. Systematic identification of DNA-binding sites and target genes of these key regulators show that they have shared and unique sets of target genes. DNA binding by MADS proteins is not based on 'simple' recognition of a specific DNA sequence, but depends on DNA structure and combinatorial interactions. Homeotic MADS proteins regulate gene expression via alternative mechanisms, one of which may be to modulate chromatin structure and accessibility in their target gene promoters.
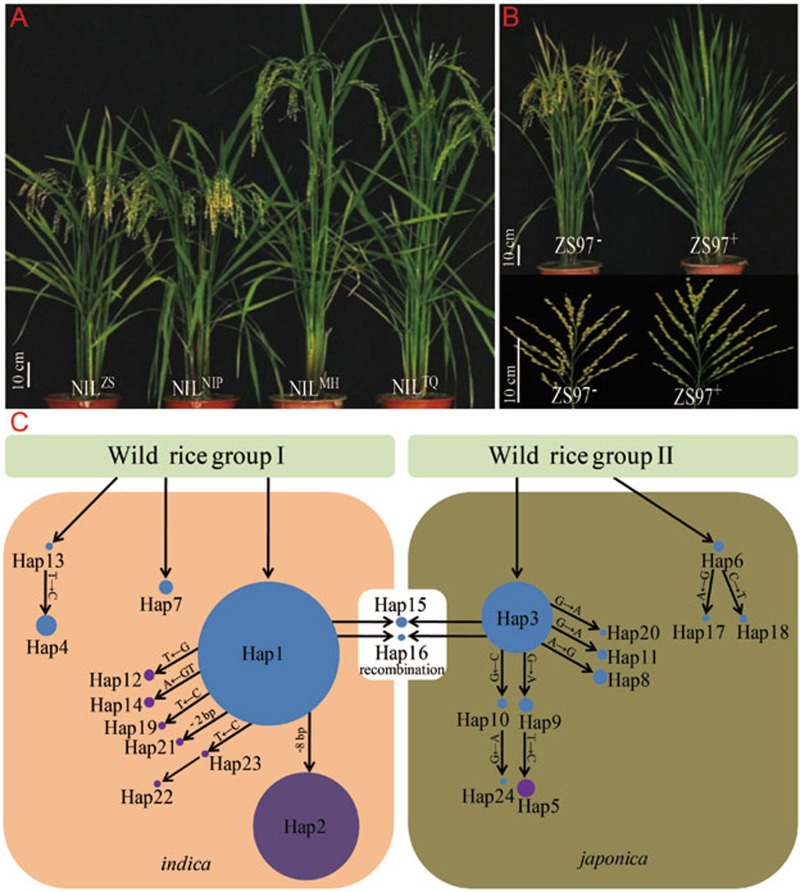
Natural variation in Ghd7.1 plays an important role in grain yield and adaptation in rice
Authors:Wenhao Yan, Haiyang Liu, Xiangchun Zhou, Qiuping Li, Jia Zhang, Li Lu, Touming Liu, Haijun Liu, Chengjun Zhang, Zhanyi Zhang, Guojing Shen, Wen Yao, Huaxia Chen, Sibin Yu, Weibo Xie, Yongzhong Xing
Journal: Cell Research
In conclusion, Ghd7.1 contributes greatly to regulating rice photoperiodic flowering, plant architecture and grain productivity. The retention of its pre-existing genetic variants in ancestral species and the acquisition of mutations after domestication together have contributed to rice adaptation. Moreover, the isolation of Ghd7.1 provides an opportunity to breed high-yield varieties with improved adaptive flexibility for special farming regions.

Molecular Diagnostics in Rice (Oryza sativa)
Authors:Wenhao Yan, Zhongmin Han & Yongzhong Xing
Journal: Diagnostics in Plant Breeding
Rice grain yield and its quality are quantitatively inherited. These traits are controlled by their genetic constitution, and environmental factors including light, temperature, fertilizers, and biotic stress. With completion of the rice genome sequence, dissection of the genetic basis of yield traits as well as rice quality advanced substantially. Hundreds of quantitative trait loci (QTL) for these traits were identified on the basis of molecular linkage maps. Dozens of disease and insect resistant QTL/genes were identified as well. Numerous markers linked to major QTL were anchored in the process of fine mapping. Map-based cloning of QTL isolated dozens of genes controlling yield components, rice quality, and biotic stress tolerance. Functional markers were developed based on these functional genes. These linked markers and functional markers started to play important roles in developing rice varieties with higher quality, higher yield potential, and greater yield stability.

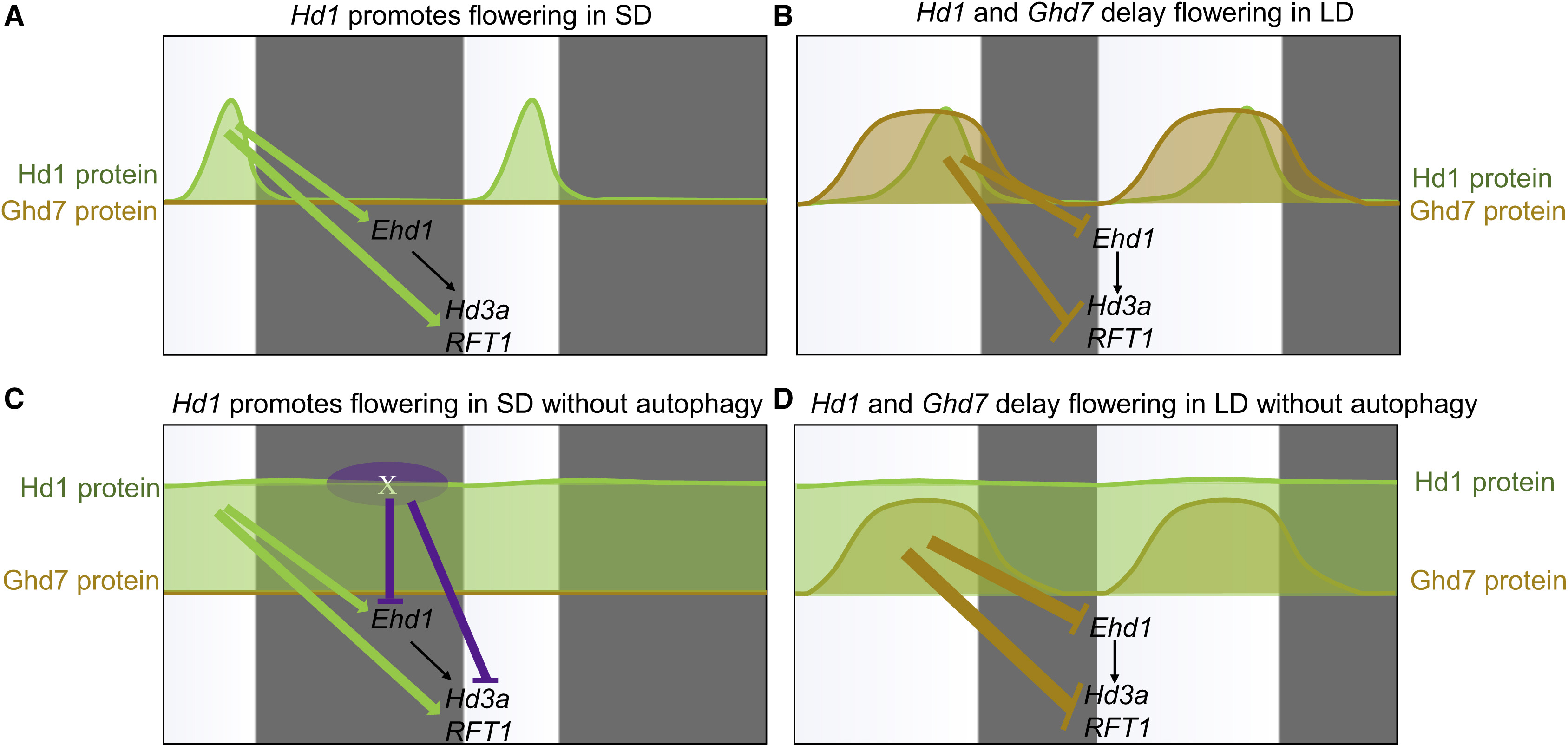
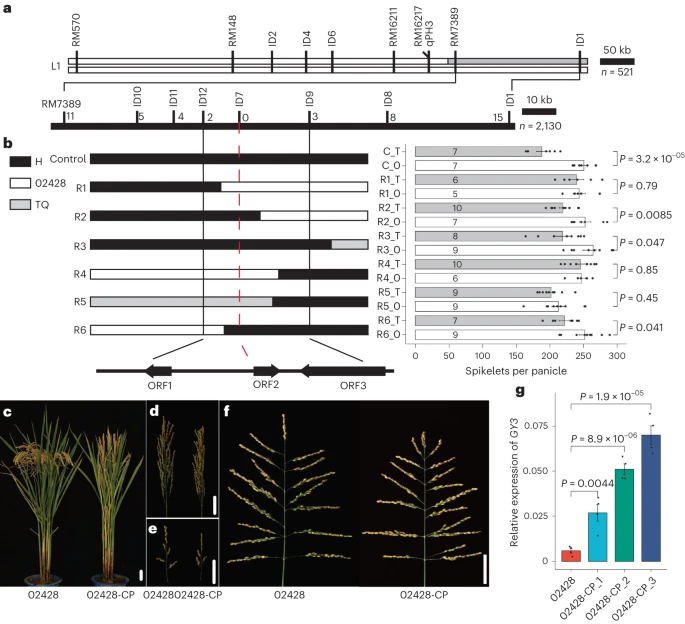
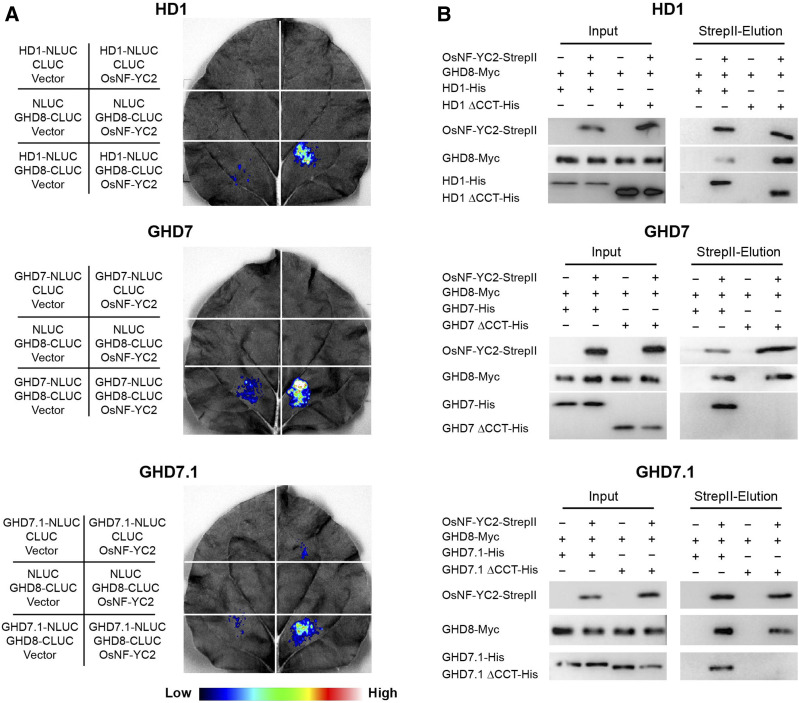
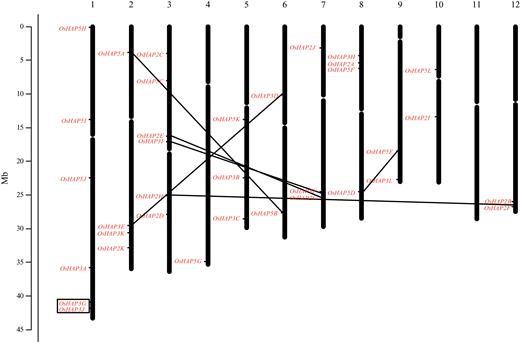
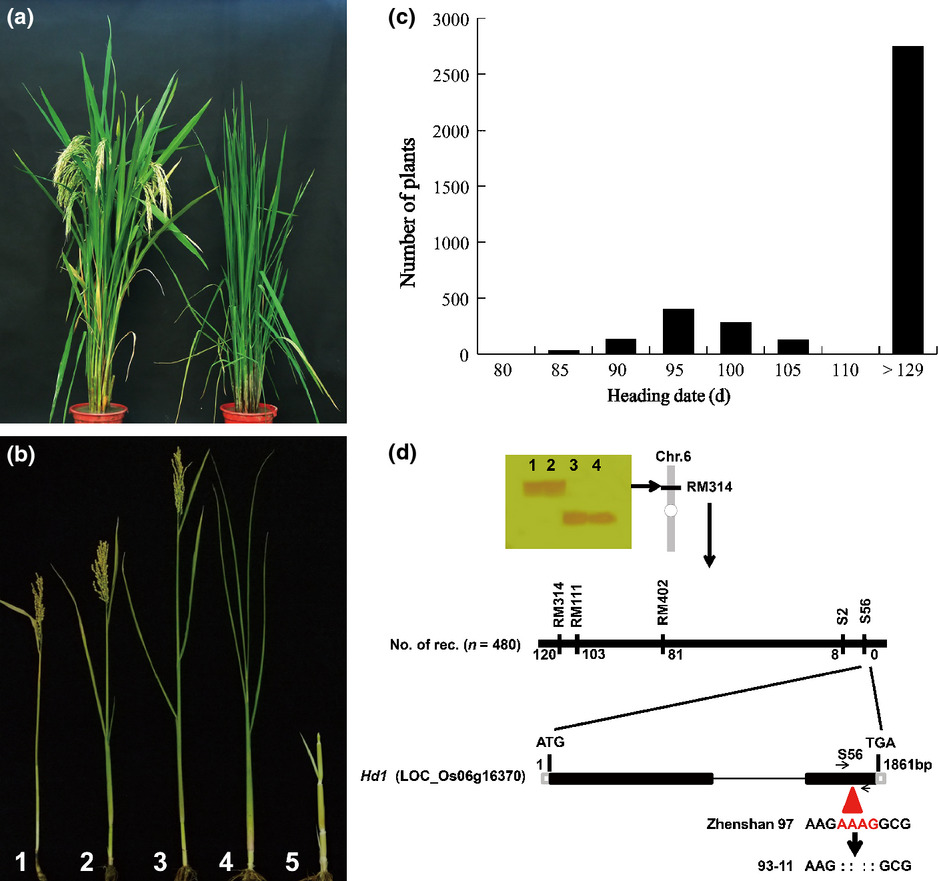
A Major QTL, Ghd8, Plays Pleiotropic Roles in Regulating Grain Productivity, Plant Height, and Heading Date in Rice
Authors:Wen-Hao Yan, Peng Wang, Hua-Xia Chen, Hong-Ju Zhou, Qiu-Ping Li, Chong-Rong Wang, Ze-Hong Ding, Yu-Shan Zhang, Si-Bin Yu, Yong-Zhong Xing, Qi-Fa Zhang
Journal: Molecular Plant
Rice yield and heading date are two distinct traits controlled by quantitative trait loci (QTLs). The dissection of molecular mechanisms underlying rice yield traits is important for developing high-yielding rice varieties. Here, we report the cloning and characterization of Ghd8, a major QTL with pleiotropic effects on grain yield, heading date, and plant height. Two sets of near isogenic line populations were developed for the cloning of Ghd8. Ghd8 was narrowed down to a 20-kb region containing two putative genes, of which one encodes the OsHAP3 subunit of a CCAAT-box binding protein (HAP complex); this gene was regarded as the Ghd8 candidate. A complementary test confirmed the identity and pleiotropic effects of the gene; interestingly, the genetic effect of Ghd8 was dependent on its genetic background. By regulating Ehd1, RFT1, and Hd3a, Ghd8 delayed flowering under long-day conditions, but promoted flowering under short-day conditions. Ghd8 up-regulated MOC1, a key gene controlling tillering and branching; this increased the number of tillers, primary and secondary branches, thus producing 50% more grains per plant. The ectopic expression of Ghd8 in Arabidopsis caused early flowering by 10 d-a situation similar to the one observed by its homolog AtHAP3b, when compared to wild-type under long-day conditions; these findings indicate the conserved function of Ghd8 and AtHAP3b in flowering in Arabidopsis. Our results demonstrated the important roles of Ghd8 in rice yield formation and flowering, as well as its opposite functions in flowering between rice and Arabidopsis under long-day conditions.